Draft a CSR — from protocol, IB, and raw datasets.
Clinical study reports take medical writing teams months per study. Most of the effort is structural: pulling boilerplate from the protocol, formatting tables derived from the trial’s datasets, threading Investigator Brochure content into safety narratives, mirroring regulatory-accepted language.
Ingests the full source material
- Protocol and amendments
- Investigator Brochure
- Raw clinical datasets: SAS transport files (
.xpt,.sas7bdat), CSVs, SDTM and ADaM domains - Prior CSRs for related studies, TFLs, subject listings
Analyzes clinical data directly
Not just assembles documents — analyzes.
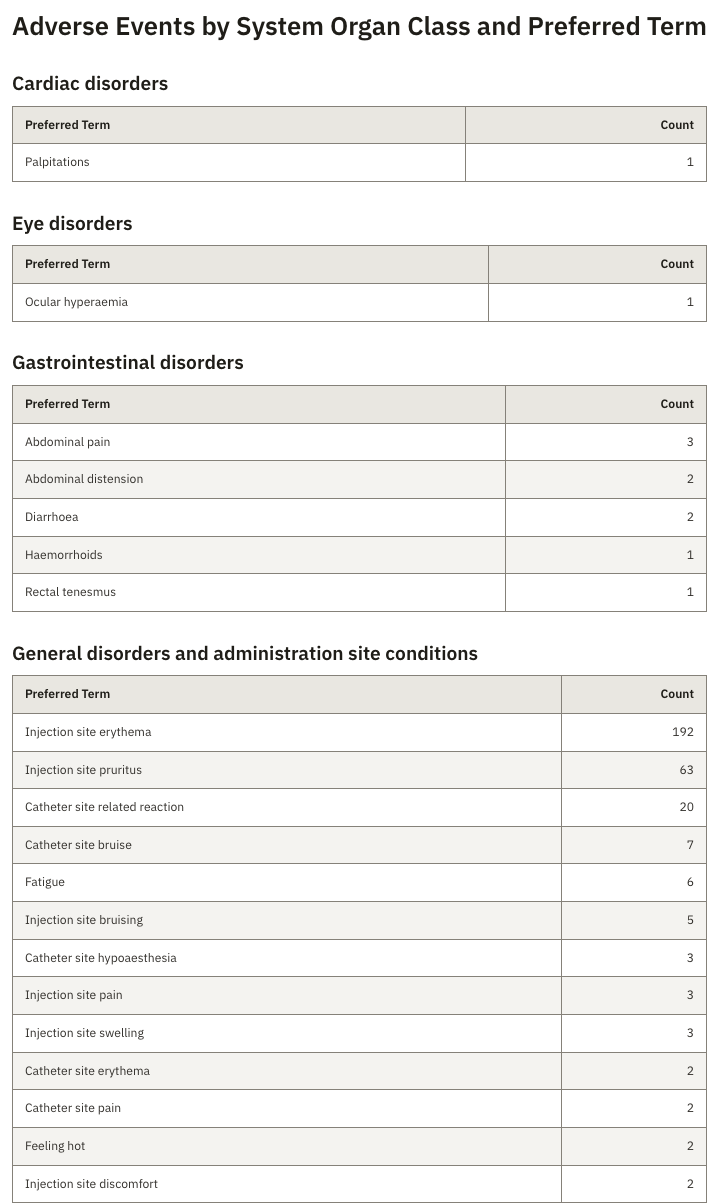
- Aggregate adverse events across arms, body systems, severity, and causality
- Generate TFL-style tables straight from SDTM and ADaM
- Draft AE narratives grounded in the source subject record
- Summarize efficacy outcomes against the protocol’s primary and secondary endpoints
- Flag data anomalies worth surfacing to the biostatistician
Drafts CSR sections to ICH E3 structure
Synopsis, Introduction, Objectives, Methods, Study Population, Efficacy Results, Safety Results, Discussion, Conclusions. Word and HTML outputs, ready for medical-writer review.
Traceable by construction
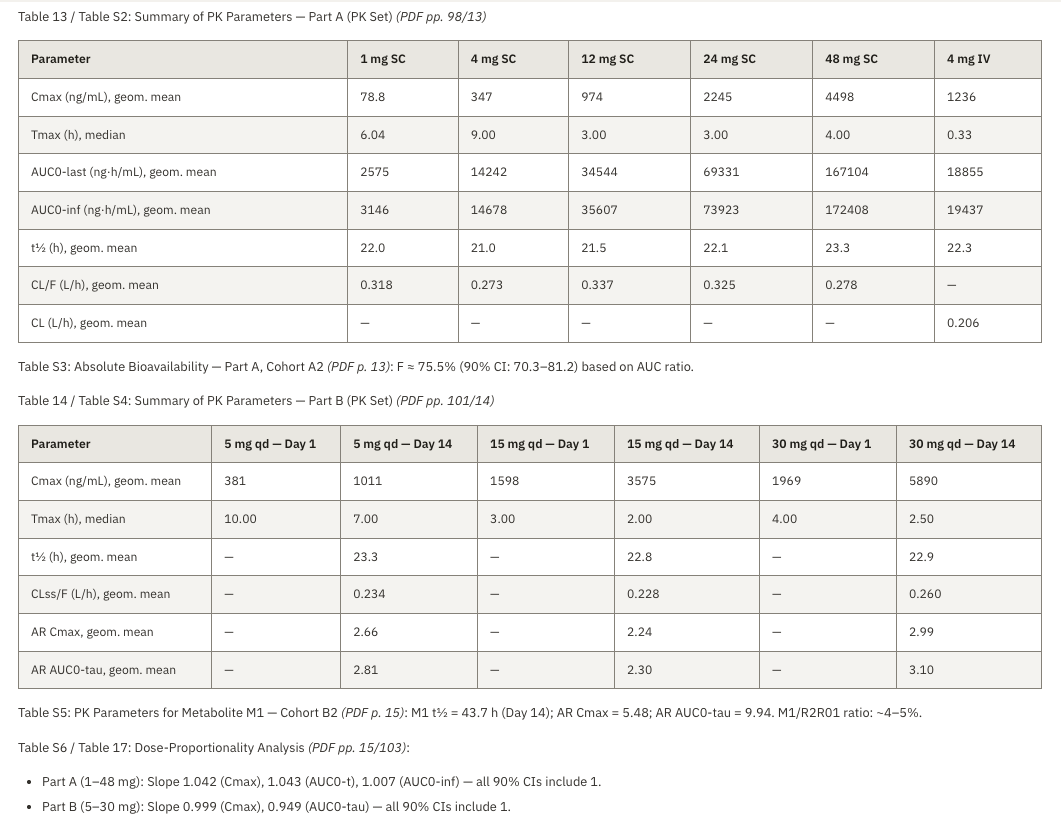
Every generated claim links back to a source document plus page, or a dataset row. QC takes minutes per section rather than hours.

Ask Vera across the dataset
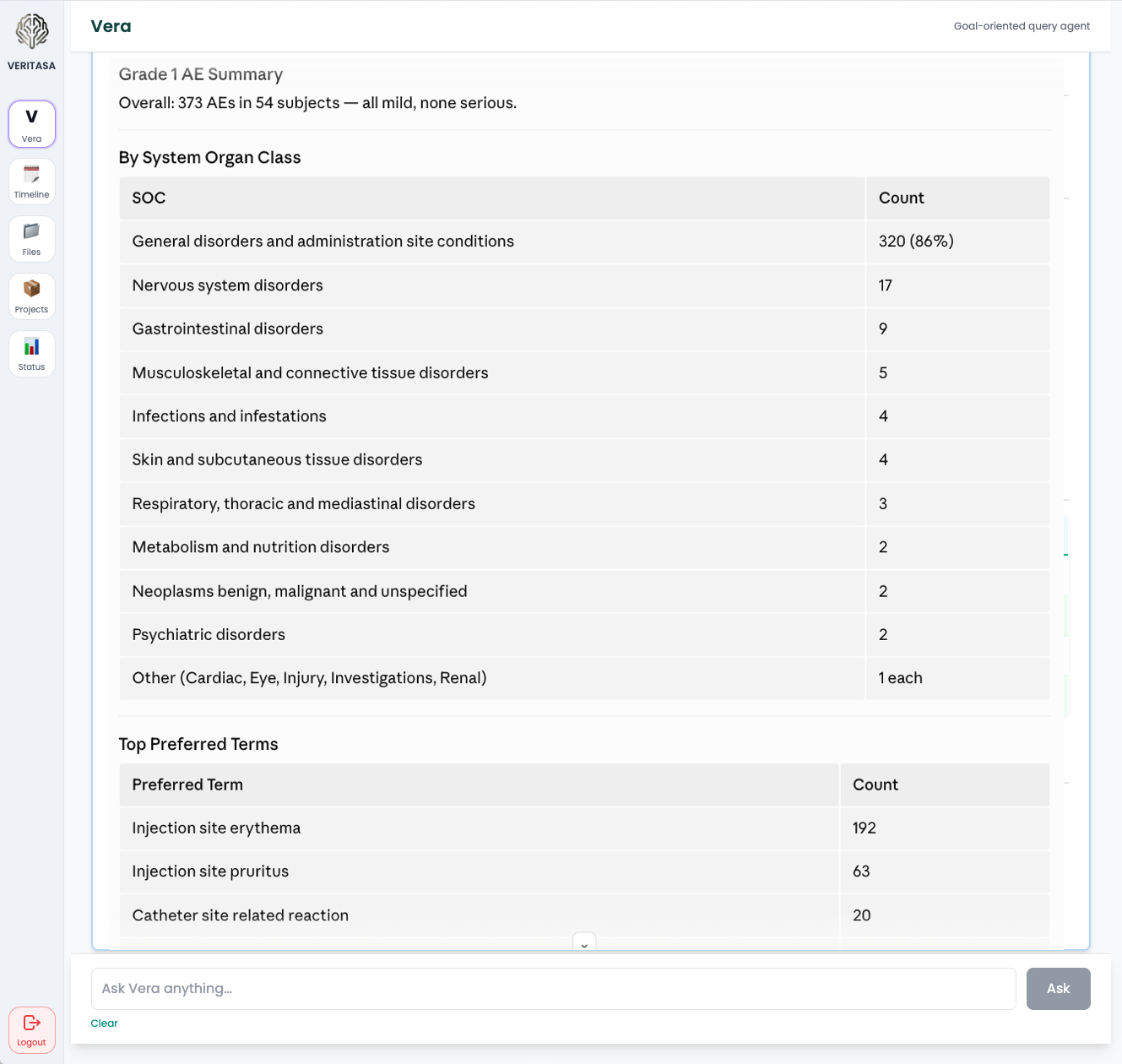
- “Summarize Grade 3+ AEs in Arm B vs. Arm C.”
- “Draft the safety narrative for subject 1045.”
- “Pull the primary endpoint table from ADaM ADSL + ADTTE.”
- “Any subjects with treatment-related SAEs outside the Investigator Brochure’s known safety profile?”
Grounded in regulatory precedent
Uses published EMA clinical data publications as a reference library of accepted CSR structure and language — so outputs resemble what regulators have already accepted.
Standards we design to — see Security.